
Is Open Source NGS Software for You?
An interview with Eric Cabot Ph.D.
 Dr. Eric Cabot received his MS from the University of British Columbia and his PhD in molecular evolutionary biology at Simon Fraser University. As a postdoc, he performed research at the National Institutes of Health and the University of Chicago. Since then he has performed biology and bioinformatics research in the private sector and in academia. Dr. Cabot’s experience includes running a bioinformatics core facility at the University of Wisconsin. He now works as a Senior Scientist and Customer Support Specialist at DNASTAR. In his over 30 years of experience, Dr. Cabot has used and supported countless bioinformatics tools, including free and commercial software applications. Here he shares some practical advice for anyone considering the merits of open source NGS analysis software.
Dr. Eric Cabot received his MS from the University of British Columbia and his PhD in molecular evolutionary biology at Simon Fraser University. As a postdoc, he performed research at the National Institutes of Health and the University of Chicago. Since then he has performed biology and bioinformatics research in the private sector and in academia. Dr. Cabot’s experience includes running a bioinformatics core facility at the University of Wisconsin. He now works as a Senior Scientist and Customer Support Specialist at DNASTAR. In his over 30 years of experience, Dr. Cabot has used and supported countless bioinformatics tools, including free and commercial software applications. Here he shares some practical advice for anyone considering the merits of open source NGS analysis software.
What are the largest obstacles that users face when analyzing next-gen sequencing data?
When choosing software for assembling next-gen data, people often focus only on accuracy and computational speed. You obviously need a lot of disk space, because you’re dealing with large amounts of data. And that often means you are going to spend a lot of time on the analysis.

However, there are additional usability factors that should be considered. For instance, are the assembly and analysis tools intuitive? How long will it take to gain expertise with the software?
With open source software, in particular, there can be a steep learning curve. By necessity, there are many different pieces of software and many steps involved. Often you don’t know if you are using the latest version of a tool, and the tools are not necessarily fast, either. Each tool has its own quirks. Putting together a workflow is a challenge because it requires a relatively high level of computer expertise.
So the biggest hurdles are the size of the data, the time and expertise involved in obtaining and using the software, and then finding a way to look at the results.
What steps are involved in setting up a pipeline using open source tools?
Before doing anything else, you need to identify all the components you need. For example, you need to obtain an alignment program, such as BWA or BOWTIE. You also need helper applications, such as SAMtools, to help manage and analyze the data. Once you’ve identified the tools you need, you may have to compile and configure them yourself.
Prior to running the alignment, most programs require you to prepare, or index, the reference genome. Then you align, or map, your reads, usually resulting in a large SAM file. Then you need to convert to the SAM file to its binary counterpart, a BAM file. Once you have a BAM file, you need to sort and index the file for downstream analysis. Finally, you have to use another tool such as the GATK HaplotypeCaller to perform variant calling, because there might be some gaps caused by insertions and deletions. Depending on the tool used for variant detection, you might also require an additional tool such as GATK’s RealignerTargetCreator to improve alignments in regions flanking insertions and deletions.
All of these steps require learning and running a variety of tools with their appropriate command line options, which can take a lot of time.
How do you learn all the command line options in the various tools?
For most public software, you have to rely on forums for guidance. Sometimes there are additional resources online, but these are often not maintained. Many people are just running one or two projects, and they don’t want to invest a lot of time learning how to use the assembly and analysis tools.
What about downstream analysis of the alignment and variant data?
Some aligners are very stringent and throw away too much data. By erring on the side of caution, these aligners end up missing some true variants. It is important to understand the limits of your alignment tool variant caller, including the sensitivity and specificity of the method used.
In addition, to make sense of your SNPs, you need to be able to distinguish known SNPs from novel SNPs. Many open source tools just provide a table of called variants and require additional scripting steps to compare the found SNPs to known SNPs.
Are there any special considerations for RNA-Seq projects?
Again, users should be mindful of additional tools and resources they may need to learn. For example, you may need to set up and learn to use R in order to utilize DESEQ2 and EdgeR analysis methods in your pipeline. Many users don’t have the time or resources to incorporate these add-ons, in which case an integrated package that utilizes these analysis methods may be preferable.
How long does it take to set up and run a pipeline like this?
Each of the individual steps and commands can take anywhere from a few minutes to several hours. But the first time you’re trying to set up the open source pipeline and connect the various pieces of software, it is often a few weeks before you can actually begin to see and analyze results.
What are some benefits to using a commercial software package for next-gen sequence alignment and analysis?
The biggest contrast with open-source software is that most of the learning curve is removed when you use a commercial software package like Lasergene Genomics. With Lasergene, you simply install software, select a genomics or transcriptomics workflow from the Navigator (shown below), and spend a couple of minutes setting up your project in SeqMan NGen’s project wizard.
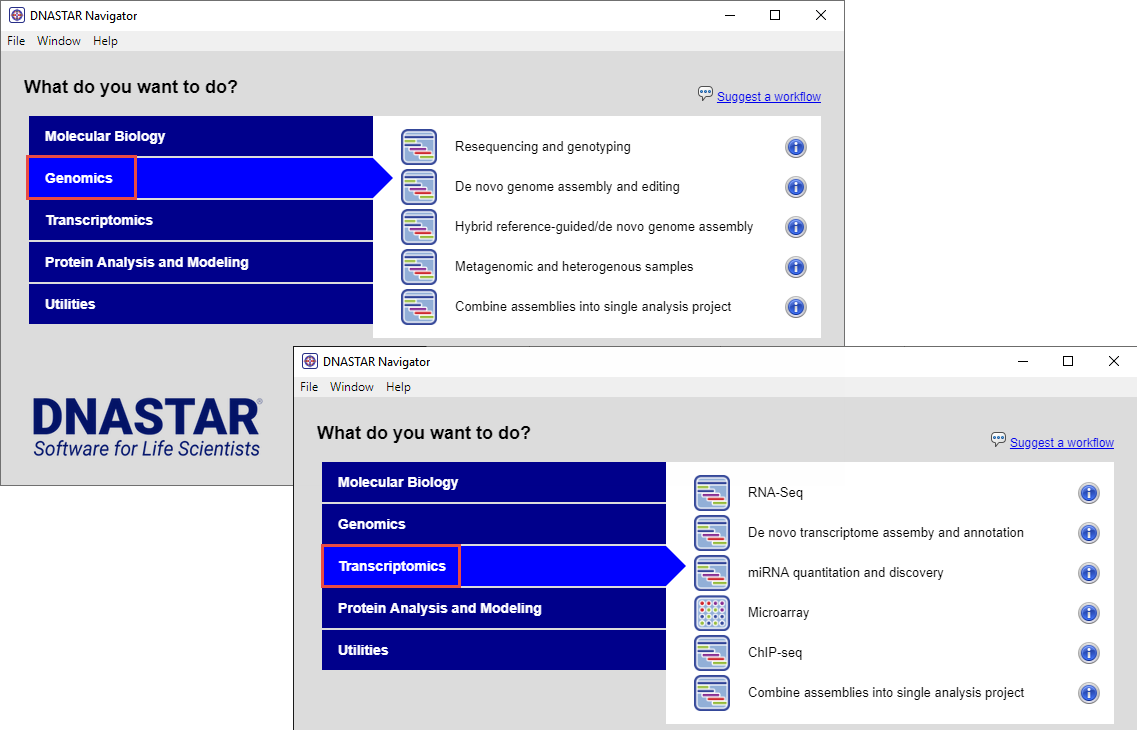 One step in the wizard lets you choose an up-to-date genome template package to download “behind the scenes” from the DNASTAR website. If you are working with RNA-Seq data, Lasergene’s ArrayStar application supports Bioconductor modules DESQ2 and EdgeR. You do not need to know anything about R; just choose the desired algorithm from a drop-down menu. Minutes later, you can start running your first assembly. If you have insufficient computer hardware, usually disk space or RAM, you can tell the wizard to run the assembly in the DNASTAR Cloud.
One step in the wizard lets you choose an up-to-date genome template package to download “behind the scenes” from the DNASTAR website. If you are working with RNA-Seq data, Lasergene’s ArrayStar application supports Bioconductor modules DESQ2 and EdgeR. You do not need to know anything about R; just choose the desired algorithm from a drop-down menu. Minutes later, you can start running your first assembly. If you have insufficient computer hardware, usually disk space or RAM, you can tell the wizard to run the assembly in the DNASTAR Cloud.
You can usually begin analyzing your results the same day. Lasergene lets you view and edit non-BAM assemblies or compare multiple BAM assemblies, including those from different workflows (e.g., ChIP-Seq vs. RNA-Seq).
Unlike open-source software, Lasergene has built-in filters to easily identify the SNPs of interest for a given project. Lasergene uses minimal hard filters during alignment but lets you easily apply additional, more stringent filters after the alignment is complete. The big benefit here is that these “soft” filters can be changed on the fly without rerunning the alignment.
Additionally, Lasergene offers a variety of integrated SNP- and gene-level annotation databases which allow you to use published clinical, population, ontology and other data as part of your filtering criteria.
**************************************************************************************
Not ready to commit to learning open source tools?
Request a free trial of Lasergene Genomics to experience the benefits of an integrated solution for NGS alignment, variant calling and downstream analysis for yourself.
Leave a Reply
Your email is safe with us.